
Vibrational Probes: From Small Molecule Solvatochromism Theory and Experiments to Applications in Complex Systems
| author | Minhaeng Cho |
|---|---|
| Homepage | http://cmsd.ibs.re.kr/html/cmsd_en/ |
| journal | Acc. Chem. Res., 2017, 50 (4), pp 968–976 |
| Attachment '1' |
|---|
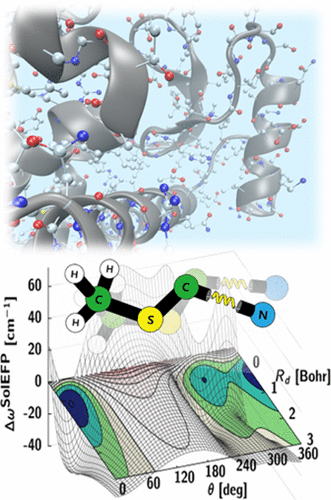
The vibrational frequency of a chosen normal mode is one of the most accurately measurable spectroscopic properties of molecules in condensed phases. Accordingly, infrared absorption and Raman scattering spectroscopy have provided valuable information on both distributions and ensemble-average values of molecular vibrational frequencies, and these frequencies are now routinely used to investigate structure, conformation, and even absolute configuration of chemical and biological molecules of interest. Recent advancements in coherent time-domain nonlinear vibrational spectroscopy have allowed the study of heterogeneous distributions of local structures and thermally driven ultrafast fluctuations of vibrational frequencies. To fully utilize IR probe functional groups for quantitative bioassays, a variety of biological and chemical techniques have been developed to site-specifically introduce vibrational probe groups into proteins and nucleic acids. These IR-probe-labeled biomolecules and chemically reactive systems are subject to linear and nonlinear vibrational spectroscopic investigations and provide information on the local electric field, conformational changes, site–site protein contacts, and/or function-defining features of biomolecules.
A rapidly expanding library of data from such experiments requires an interpretive method with atom-level chemical accuracy. However, despite prolonged efforts to develop an all-encompassing theory for describing vibrational solvatochromism and electrochromism as well as dynamic fluctuations of instantaneous vibrational frequencies, purely empirical and highly approximate theoretical models have often been used to interpret experimental results. They are, in many cases, based on the simple assumption that the vibrational frequency of an IR reporter is solely dictated by electric potential or field distribution around the vibrational chromophore. Such simplified description of vibrational solvatochromism generally referred to as vibrational Stark effect theory has been considered to be quite appealing and, even in some cases, e.g., carbonyl stretch modes in amide, ester, ketone, and carbonate compounds or proteins, it works quantitatively well, which makes it highly useful in determining the strength of local electric field around the IR chromophore. However, noting that the vibrational frequency shift results from changes of solute–solvent intermolecular interaction potential along its normal coordinate, Pauli exclusion repulsion, polarization, charge transfer, and dispersion interactions, in addition to the electrostatic interaction between distributed charges of both vibrational chromophore and solvent molecules, are to be properly included in the theoretical description of vibrational solvatochromism. Since the electrostatic and nonelectrostatic intermolecular interaction components have distinctively different distance and orientation dependences, they affect the solvatochromic vibrational properties in a completely different manner.
Over the past few years, we have developed a systematic approach to simulating vibrational solvatochromic data based on the effective fragment potential approach, one of the most accurate and rigorous theories on intermolecular interactions. We have further elucidated the interplay of local electric field with the general vibrational solvatochromism of small IR probes in either solvents or complicated biological systems, with emphasis on contributions from non-Coulombic intermolecular interactions to vibrational frequency shifts and fluctuations. With its rigorous foundation and close relation to quantitative interpretation of experimental data, this and related theoretical approaches and experiments will be of use in studying and quantifying the structure and dynamics of biomolecules with unprecedented time and spatial resolution when combined with time-resolved vibrational spectroscopy and chemically sensitive vibrational imaging techniques.
-
Read More

Chemical Fields: Directing Atom Migration in the Multiphasic Nanocrystal
Kwangyeol Leehttp://nanolab.korea.ac.kr/Atoms in a bulk solid phase are usually trapped to fixed positions and can change their position only under certain conditions (e.g., at a melting point) due to the high energy barrier of migration between positions within the crystal lattic...Date2023.05.08 Bywebmaster2 Views35 -
Read More
Electronic Mechanism of In Situ Inversion of Rectification Polarity in Supramolecular Engineered Monalayer
Hyo Jae Yoonhttps://hyojaeyoon.wixsite.com/ommlThis paper describes polarity inversion in molecular rectification and the related mechanism. Using supramolecular engineered, ultrastable binary mixed self-assembled monolayer (SAM) composed of organic molecular diode (SC11BIPY) and inert r...Date2023.05.08 Bywebmaster2 Views39 -
Read More
Direct observation of protein structural transitions through entire amyloid aggregation processes in water using 2D-IR spectroscopy
Hugh I. Kim, Kyungwon Kwakhttps://kkwaklab.wixsite.com/quantspeclabAmyloid proteins that undergo self-assembly to form insoluble fibrillar aggregates have attracted much attention due to their role in biological and pathological significance in amyloidosis. This study aims to understand the amyloid aggregat...Date2023.05.08 Bywebmaster2 Views35 -
Read More
Harnessing GLUT1-Targeted Pro-oxidant Ascorbate for Synergistic Phototherapeutics
Jong Seung Kimhttp://orgchem.korea.ac.krDespite extensive efforts to realize effective photodynamic therapy (PDT), there is still a lack of therapeutic approaches concisely structured to mitigate the major obstacles of PDT in clinical applications. Herein, we report a molecular st...Date2023.05.08 Bywebmaster2 Views35 -
Read More
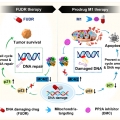
DNA-Damage-Response-Targeting Mitochondri-Activated Multifunctional Prodrug Strategy for Self-Denfensive Tumor Therapy
Jong Seung Kimhttp://orgchem.korea.ac.kr/We report a novel multifunctional construct, M1, designed explicitly to target the DNA damage response in cancer cells. M1 contains both a floxuridine (FUDR) and protein phosphatase 2A (PP2A) inhibitor combined with a GSH-sensitive linker. F...Date2023.05.04 Bywebmaster2 Views35 -
Read More
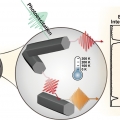
Midwavelength Infrared Colloidal Nanowire Laser
Kwang Seob Jeonghttps://kwangsjeong.wixsite.com/ksjlab-koreaunivRealizing bright colloidal infrared emitters in the midwavelength infrared (or mid-IR), which can be used for low-power IR light-emitting diodes (LEDs), sensors, and deep-tissue imaging, has been a challenge for the last few decades. Here, w...Date2023.05.04 Bywebmaster2 Views37 -
Read More
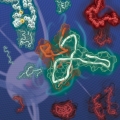
Kinetic Modulation of Amyloid-β (1-42) Aggegation and Toxicity by Structure-Based Rational Design
Hugh I. Kimhttps://www.hughkimlab.com/Several point mutations can modulate protein structure and dynamics, leading to different natures. Especially in the case of amyloidogenic proteins closely related to neurodegenerative diseases, structural changes originating from point muta...Date2023.05.04 Bywebmaster2 Views78 -
Read More
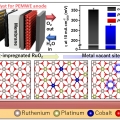
Safeguarding the RuO2 phase against lattice oxygen oxidation during acidic water electrooxidation
Kwangyeol Leehttp://nanolab.korea.ac.kr/Defective RuO2 possesses excellent initial activity toward the oxygen evolution reaction in acidic water electrooxidation due to the involvement of lattice oxygens, which, however, is the very reason for the accelerated dissolution of Ru spe...Date2023.05.04 Bywebmaster2 Views36 -
Read More
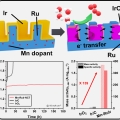
Mn-dopant differentiating the Ru and Ir oxidation states in catalytic oxides toward durable oxygen evolution reaction in acidic electrolyte
Kwangyeol Leehttp://nanolab.korea.ac.kr/Designing an efficient and durable electrocatalyst for the sluggish oxygen evolution reaction (OER) at the anode remains the foremost challenge in developing proton exchange membrane (PEM) electrolyzers. Here we report a highly active and du...Date2023.05.04 Bywebmaster2 Views58 -
Read More

Dynamic Water Promotes Lithium-Ion Transport in Superconcentrated and Eutectic Aqueous Electrolytes
Kyungwon Kwak, Minhaeng Chohttps://kkwaklab.wixsite.com/quantspeclabSuperconcentrated aqueous electrolytes have shown promise as safe and high-voltage lithium-ion battery (LIB) electrolytes. However, the interplay of lithium-ion solvation structure and dynamics with fast Li-ion transport has not been elucida...Date2023.05.04 Bywebmaster2 Views39 -
Read More
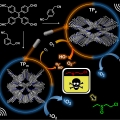
Isomeric sp2-C conjugated Porous Organic Polymer-mediated Photo- and Sono-catalytic Detoxification of Sulfur Mustard Simulant under Ambient Conditions
Jong Seung Kim, Chang Seop Honghttp://MatterThe development of efficient strategies for the sustainable detoxification of mustard gas simulants has longstanding demand for human safety. Here, we present for the first time a photo- and sono-catalyzed selective detoxification of mustard...Date2023.05.04 Bywebmaster2 Views39 -
Read More

Vertical-crystalline Fe-doped β-Ni oxyhydroxides for highly active and stable oxygen evolution reaction
Kwangyeol Leehttp://nanolab.korea.ac.kr/The layered transition metal oxyhydroxides have received increasing interest owing to the efficient energy conversion performance and material stability during the oxygen evolution reaction (OER). In particular, Fe-doped NiOOH has shown reco...Date2023.05.04 Bywebmaster2 Views41 -
Read More
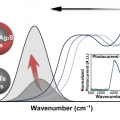
Extended Short-Wavelength Infrared Photoluminescence and Photocurrent of Nonstoichiometric Silver Telluride Collodial Nanocrystals
Kwang Seob Jeonghttps://kwangsjeong.wixsite.com/ksjlab-koreaunivDemands on nontoxic nanomaterials in the short-wavelength infrared (SWIR) have rapidly grown over the past decade. Here, we present the nonstoichiometric silver chalcogenide nanocrystals of AgxTe (x > 2) and Ag2Te/Ag2S CQDs with a tunable ba...Date2023.05.04 Bywebmaster2 Views447 -
Read More
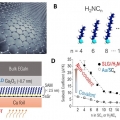
Enhanced Thermopower of Saturated Molecules by Noncovalent Anchor-Induced Electron Doping of Single-Layer Graphene Electrode
Hyo Jae Yoonhttps://hyojaeyoon.wixsite.com/ommlEnhancing thermopower is a key goal in organic and molecular thermoelectrics. Herein, it is shown that introducing noncovalent contact with a single-layer graphene (SLG) electrode improves the thermopower of saturated molecules as compared t...Date2023.05.04 Bywebmaster2 Views31 -
Read More
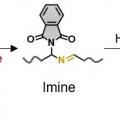
Mechanical Force for the Transformation of Aziridine into Imine
Hyo Jae Yoonhttps://hyojaeyoon.wixsite.com/ommlForce-selective mechanochemical reactions may be important for applications in polymer mechanochemistry, yet it is difficult to achieve such reactions. This paper reports that cis-N-phthalimidoaziridine incorporated into a macromolecular bac...Date2023.05.04 Bywebmaster2 Views27 -
Read More
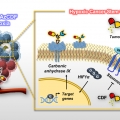
A Small Molecule Strategy for Targeting Cancer Stem Cells in Hypoxic Microenvironments and Preventing Tumorigenesis
Jong Seung Kimhttp://orgchem.korea.ac.kr/Breast cancer consists of heterogenic subpopulations, which determine the prognosis and response to chemotherapy. Among these subpopulations, a very limited number of cancer cells are particularly problematic. These cells, known as breast ca...Date2023.05.04 Bywebmaster2 Views24 -
Read More
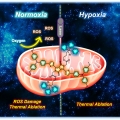
Mitochondria-targeted nanotheranostic: Harnessing single-laser-activated dual phototherapeutic processing for hypoxic tumor treatment
Jong Seung Kimhttp://orgchem.korea.ac.kr/Realizing maximum tumor suppression along with preventing tumor regrowth by optimizing the photon usage in phototherapy remains a major challenge. Herein, a mitochondria-targeted phototheranostic nanoformulation (MsPDTT NPs) was prepared fro...Date2023.05.04 Bywebmaster2 Views24 -
Read More
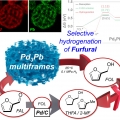
Pb3Pb Nanosponges for Selective Conversion of Furfural to Furfuryl Alcohol under Mild Condition
Heejin Kim, Kwangyeol Leehttp://nanolab.korea.ac.kr/Alloy structures with high catalytic surface areas and tunable surface energies can lead to high catalytic selectivity and activities. Herein, the synthesis of sponge-like Pd3Pb multiframes (Pd3Pb MFs) is reported by using the thermodynamica...Date2023.05.04 Bywebmaster2 Views136 -
Read More

Deep Learning Optical Spectroscopy Based on Experimental Database: Potential Applications to Molecular Design
Sungnam Parkhttps://ultrafastspec.wixsite.com/sparkAccurate and reliable prediction of the optical and photophysical properties of organic compounds is important in various research fields. Here, we developed deep learning (DL) optical spectroscopy using a DL model and experimental database ...Date2023.05.04 Bywebmaster2 Views29 -
Read More

Wettability of graphene and interfacial water structure
Kyungwon Kwak, Minhaeng Chohttps://cmsd.ibs.re.kr/html/cmsd_en/Understanding the wettability of graphene is important for various applications, such as water desalination, energy storage, and catalysis. However, the detailed water hydrogen-bonding structure at the water-graphene interface is not yet ful...Date2023.05.04 Bywebmaster2 Views25
Designed by sketchbooks.co.kr / sketchbook5 board skin