
Multiplexed Post-Experimental Monoisotopic Mass Refinement (mPE-MMR) to Increase Sensitivity and Accuracy in Peptide Identifications from Tandem Mass Spectra of Cofragmentation
| author | Sang-Won Lee |
|---|---|
| Homepage | http://ionchem.korea.ac.kr/ |
| journal | Anal. Chem., 2017, 89 (2), pp 1244–1253 |
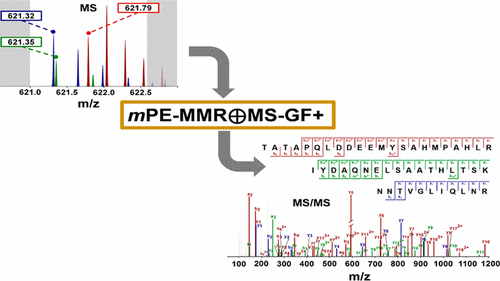
Mass spectrometry (MS)-based proteomics, which uses high-resolution hybrid mass spectrometers such as the quadrupole-orbitrap mass spectrometer, can yield tens of thousands of tandem mass (MS/MS) spectra of high resolution during a routine bottom-up experiment. Despite being a fundamental and key step in MS-based proteomics, the accurate determination and assignment of precursor monoisotopic masses to the MS/MS spectra remains difficult. The difficulties stem from imperfect isotopic envelopes of precursor ions, inaccurate charge states for precursor ions, and cofragmentation. We describe a composite method of utilizing MS data to assign accurate monoisotopic masses to MS/MS spectra, including those subject to cofragmentation. The method, “multiplexed post-experiment monoisotopic mass refinement” (mPE-MMR), consists of the following: multiplexing of precursor masses to assign multiple monoisotopic masses of cofragmented peptides to the corresponding multiplexed MS/MS spectra, multiplexing of charge states to assign correct charges to the precursor ions of MS/MS spectra with no charge information, and mass correction for inaccurate monoisotopic peak picking. When combined with MS-GF+, a database search algorithm based on fragment mass difference, mPE-MMR effectively increases both sensitivity and accuracy in peptide identification from complex high-throughput proteomics data compared to conventional methods.
http://pubs.acs.org/doi/abs/10.1021/acs.analchem.6b03874
« Prev Molecular design of a wide-band-gap conjugated polymer for ef...
 Molecular design of a wide-band-gap conjugated polymer for ef...
2017.03.31by Manager
〈
Molecular design of a wide-band-gap conjugated polymer for ef...
2017.03.31by Manager
〈
Key Structural Elements for Cellular Uptake of Acinetobactin,... Next »
 Key Structural Elements for Cellular Uptake of Acinetobactin,...
2017.02.07by Manager
〉
Key Structural Elements for Cellular Uptake of Acinetobactin,...
2017.02.07by Manager
〉
-
Read More
Cell imaging: An intracellular dance visualized
Sang-Hee Shimhttp://Nature 546, 39–40 (doi:10.1038/nature22500)The development of a microscopy technique that enables observation of the interactions between six types of organelle, in 3D and over time, holds promise for improving our understanding of intracellular processes. See Letter p.162 http://ww...Date2017.06.27 ByManager Views3855 -
Read More
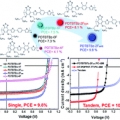
High-efficiency photovoltaic cells with wide optical band gap polymers based on fluorinated phenylene-alkoxybenzothiadiazole
Han Young Woohttp://www.ooml.korea.ac.kr/A series of semi-crystalline, wide band gap (WBG) photovoltaic polymers were synthesized with varying number and topology of fluorine substituents. To decrease intramolecular charge transfer and to modulate the resulting band gap of D–...Date2017.06.12 ByManager Views1594 -
Read More
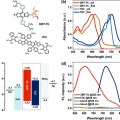
Eco-Friendly Solvent-Processed Fullerene-Free Polymer Solar Cells with over 9.7% Efficiency and Long-Term Performance Stability
Dong Hoon Choihttp://fpl.korea.ac.kr/index2.aspA wide-bandgap polymer, (poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(2,5-(methyl thiophene carboxylate))]) (3MT-Th), is synthesized to obtain a complementary broad range absorption when harmo...Date2017.06.12 ByManager Views4075 -
Read More

Iridium-Based Multimetallic Nanoframe@Nanoframe Structure: An Efficient and Robust Electrocatalyst toward Oxygen Evolution Reaction
Kwangyeol Leehttp://nanolab.korea.ac.kr/Nanoframe electrocatalysts have attracted a great interest due to their inherently high active surface area per a given mass. Although recent progress has enabled the preparation of single nanoframe structures with a variety of morphologies,...Date2017.06.12 ByManager Views1312 -
Read More
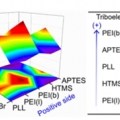
Formation of Triboelectric Series via Atomic Level Surface Functionalization for Triboelectric Energy Harvesting
Hyo Jae Yoonhttp://sites.google.com/site/ommlatkuTriboelectric charging involves frictional contact of two different materials, and their contact electrification usually relies on polarity difference in the triboelectric series. This limits the choices of materials for triboelectric contac...Date2017.05.31 ByManager Views4100 -
Read More
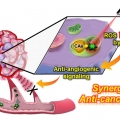
Overcoming the Limits of Hypoxia in Photodynamic Therapy: A Carbonic Anhydrase IX-Targeted Approach
Jong Seung Kimhttp://orgchem.korea.ac.kr/index2.aspA major challenge in photodynamic cancer therapy (PDT) is avoiding PDT-induced hypoxia, which can lead to cancer recurrence and progression through activation of various angiogenic factors and significantly reduce treatment outcomes. Reporte...Date2017.05.10 ByManager Views2549 -
Read More
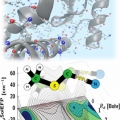
Vibrational Probes: From Small Molecule Solvatochromism Theory and Experiments to Applications in Complex Systems
Minhaeng Chohttp://cmsd.ibs.re.kr/html/cmsd_en/The vibrational frequency of a chosen normal mode is one of the most accurately measurable spectroscopic properties of molecules in condensed phases. Accordingly, infrared absorption and Raman scattering spectroscopy have provided valuable ...Date2017.04.26 ByManager Views1820 -
Read More
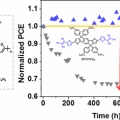
Excellent Long-Term Stability of Power Conversion Efficiency in Non-Fullerene-Based Polymer Solar Cells Bearing Tricyanovinylene-Functionalized n-Type Small Molecules
Dong Hoon Choihttp://fpl.korea.ac.kr/index2.aspNew small molecules having modified acceptor strength and π-conjugation length and containing dicyanovinylene (DCV) and tricyanovinylene (TCV) as a strongly electron-accepting unit with indacenodithiophene, IDT(DCV)2, IDT(TCV)2, and IDTT(...Date2017.04.06 ByManager Views10973 -
Read More
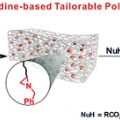
Aziridine-Functionalized Polydimethylsiloxanes for Tailorable Polymeric Scaffolds: Aziridine as a Clickable Moiety for Structural Modification of Materials
Hyo Jae Yoonhttps://sites.google.com/site/ommlatku/This paper shows that tailorable polymeric scaffolds on a molecular scale could be achieved through the ring opening reaction of a three-membered N-heterocyclic compound, aziridine. Aziridine is incorporated into an elastomeric polymer backb...Date2017.04.06 ByManager Views1920 -
Read More
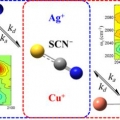
Effect of ion-ligand binding on ion pairing dynamics studied by two-dimensional infrared spectroscopy
Sungnam Parkhttp://ultrafastspec.wixsite.com/sparkCation-specific ion pairing dynamics between M+ (M=Ag or Cu) and SCN−in N,N-dimethylthioformamide (DMTF) are studied by vibrationally probing the nitrile (CN) stretching vibration. SCN− ion, which is an ambidentate ligand, readil...Date2017.04.06 ByManager Views1851 -
Read More
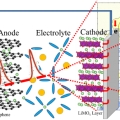
Ultrafast fluxional exchange dynamics in electrolyte solvation sheath of lithium ion battery
Kyungwon Kwak & Minhaeng Chohttp://cmsd.ibs.re.kr/html/cmsd_en/Lithium cation is the charge carrier in lithium-ion battery. Electrolyte solution in lithium-ion battery is usually based on mixed solvents consisting of polar carbonates with different aliphatic chains. Despite various experimental evidence...Date2017.03.31 ByManager Views1832 -
Read More
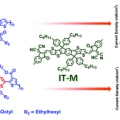
Molecular design of a wide-band-gap conjugated polymer for efficient fullerene-free polymer solar cells
Han Young Woohttp://www.ooml.korea.ac.kr/Two p-type conjugated polymers with disparate optical and electronic properties, PB3T and PB2T, were developed and applied in fullerene-free polymer solar cells (PSCs). The photovoltaic performance of the PB3T-based PSC device processed by a...Date2017.03.31 ByManager Views1844 -
Read More

Multiplexed Post-Experimental Monoisotopic Mass Refinement (mPE-MMR) to Increase Sensitivity and Accuracy in Peptide Identifications from Tandem Mass Spectra of Cofragmentation
Sang-Won Leehttp://ionchem.korea.ac.kr/Mass spectrometry (MS)-based proteomics, which uses high-resolution hybrid mass spectrometers such as the quadrupole-orbitrap mass spectrometer, can yield tens of thousands of tandem mass (MS/MS) spectra of high resolution during a routine b...Date2017.03.31 ByManager Views3858 -
Read More
Key Structural Elements for Cellular Uptake of Acinetobactin, a Major Siderophore of Acinetobacter baumannii
Hak Joong Kimhttps://sites.google.com/site/kubccb/Acinetobactin is a major siderophore utilized by the human pathogen Acinetobacter baumannii. The rapid acquisition of drug resistance by A. baumannii has garnered concern globally. Herein, acinetobactin and systematically generated analogues...Date2017.02.07 ByManager Views2375 -
Read More
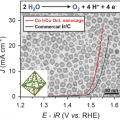
Cobalt Assisted Synthesis of IrCu Hollow Octahedral Nanocages as Highly Active Electrocatalysts toward Oxygen Evolution Reaction
Kwangyeol Leehttp://nanolab.korea.ac.kr/Development of oxygen evolution reaction (OER) catalysts with reduced precious metal content while enhancing catalytic performance has been of pivotal importance in cost-effective design of acid polymer electrolyte membrane water electrolyze...Date2017.02.07 ByManager Views1535 -
Read More
Singly and Doubly Occupied Higher Quantum State in Nanocrystals
Kwang Seob Jeonghttps://sites.google.com/site/ksjkulab/Filling the lowest quantum state of the conduction band (CB) of colloidal nanocrystals with a single electron, which is analogous to the filling the lowest unoccupied molecular orbital (LUMO) in a molecule with a single electron, has attract...Date2017.01.24 ByManager Views1786 -
Read More
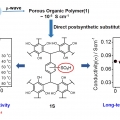
Cost-Effective, High-Performance Porous-Organic-Polymer Conductors Functionalized with Sulfonic Acid Groups by Direct Postsynthetic Substitution
Chang Seop Honghttp://immlab.korea.ac.kr/We demonstrate the facile microwave-assisted synthesis of a porous organic framework 1 and the sulfonated solid (1S) through postsubstitution. Remarkably, the conductivity of 1S showed an approximately 300-fold enhancement at 30 °...Date2016.12.22 ByManager Views1919 -
Read More
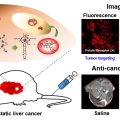
Liposomal Texaphyrin Theranostics for Metastatic Liver Cancer
Jong Seung Kimhttp://orgchem.korea.ac.kr/index2.aspReported here is a new theranostic agent, 1, which consists of a Gd3+-texaphyrin core conjugated to a doxorubicin prodrug via a disulfide bond. Conjugate 1 was designed to undergo cleavage in the presence of glutathione (GSH), a species typi...Date2016.12.21 ByManager Views2939 -
Read More
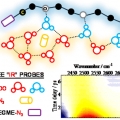
Water Dynamics in Cytoplasm-Like Crowded Environment Correlates with the Conformational Transition of the Macromolecular Crowder
Minhaeng Chohttp://cmsd.ibs.re.kr/html/cmsd_en/Polyethylene glycol (PEG) is a unique polymer material with enormous applicability in many industrial and scientific fields. Here, its use as macromolecular crowder to mimic the cellular environment in vitro is the focus of the present study...Date2016.12.16 ByManager Views6006 -
Read More
Plasmon Enhanced Direct Bandgap Emissions in Cu7S4@Au2S@Au Nanorings
Kwangyeol Leehttp://nanolab.korea.ac.kr/Nanostructured copper sulfides, promising earth-abundant p-type semiconductors, have found applications in a wide range of fields due to their versatility, tunable low bandgap, and environmental sustainability. The synthesis of hexagonal Cu7...Date2016.09.29 ByManager Views1921
Designed by sketchbooks.co.kr / sketchbook5 board skin